Note that this workflow is continually being updated. If you want to use the below commands be sure to keep track of them locally.
Last updated: 9 Oct 2018 (see revisions above for earlier minor versions and Metagenomics SOPs on the SOP for earlier major versions)
Important points to keep in mind:
The below options are not necessarily best for your data; it is important to explore different options, especially at the read quality filtering stage.
If you are confused about a particular step be sure to check out the pages under Metagenomic Resources on the right side-bar.
You should check that only the FASTQs of interest are being specified at each step (e.g. with the ls command). If you are re-running commands multiple times or using optional steps the input filenames and/or folders could differ.
The tool parallel comes up several times in this workflow. Be sure to check out our tutorial on this tool here.
You can always run parallel with the --dry-run option to see what commands are being run to double-check they are doing what you want!
This workflow starts with raw paired-end MiSeq or NextSeq data in demultiplexed FASTQ format assumed to be located within a folder called raw_data.
1. First Steps
1.1 Join lanes together (all Illumina, except MiSeq)
Concatenate multiple lanes of sequencing together (e.g. for NextSeq data). If you do NOT do this step, remember to change cat_lanes to raw_data in the further commands below that have assumed you are working with the most common type of metagenome data.
concat_lanes.pl raw_data/* -o cat_lanes -p 4
1.2 Inspect read quality
Run fastqc to allow manual inspection of the quality of sequences.
Stitch paired-end reads together (summary of stitching results are written to “pear_summary_log.txt”). Note: it is important to check the % of reads assembled. It may be better to concatenate the forward and reverse reads together if the assembly % is too low (see step 2.2).
run_pear.pl -p 4 -o stitched_reads cat_lanes/*
If you don’t stitch your reads together at this step you will need to unzip your FASTQ files before continuing with the below commands.
2. Read Quality-Control and Contaminant Screens
2.1 Running KneadData
Use kneaddata to run pre-processing tools. First Trimmomatic is run to remove low quality sequences. Then Bowtie2 is run to screen out contaminant sequences. Below we are screening out reads that map to the human or PhiX genomes. Note KneadData is being run below on all unstitched FASTQ pairs with parallel, you can see our quick tutorial on this tool here. For a detailed breakdown of the options in the below command see this page. The forward and reverse reads will be specified by “_1″ and “_2″ in the output files, ignore the “R1″ in each filename. Note that the \ characters at the end of each line are just to split the command over multiple lines to make it easier to read.
2.2 Concatenate unstitched output (Omit if data stitched above)
If you did not stitch your paired-end reads together with pear, then you can concatenate the forward and reverse FASTQs together now. Note this is done after quality filtering so that both reads in a pair are either discarded or retained. It’s important to only specify the “paired” FASTQs outputted by kneaddata in the below command.
You should check over the commands that are printed to screen to make sure the correct FASTQs are being concatenated together.
3. Determine Functions with HUMAnN2
3.1 Run HUMAnN2
Run humann2 with parallel to calculate abundance of UniRef90 gene families and MetaCyc pathways. If you are processing environment data (e.g. soil samples) the vast majority of the reads may not map using this approach. Instead, you can try mapping against the UniRef50 database (which you can point to with the --protein-database option).
Convert unstratified HUMAnN2 abundance tables to STAMP format by changing header-line. These commands remove the comment character and the spaces in the name of the first column. Trailing descriptions of the abundance datatype are also removed from each sample’s column name.
sed 's/_Abundance-RPKs//g' humann2_final_out/humann2_genefamilies_cpm_unstratified.tsv | sed 's/# Gene Family/GeneFamily/' > humann2_final_out/humann2_genefamilies_cpm_unstratified.spf
sed 's/_Abundance//g' humann2_final_out/humann2_pathabundance_cpm_unstratified.tsv | sed 's/# Pathway/Pathway/' > humann2_final_out/humann2_pathabundance_cpm_unstratified.spf
sed 's/_Coverage//g' humann2_final_out/humann2_pathcoverage_unstratified.tsv | sed 's/# Pathway/Pathway/' > humann2_final_out/humann2_pathcoverage_unstratified.spf
3.6 Extract MetaPhlAn2 taxonomic compositions
Since HUMAnN2 also runs MetaPhlAn2 as an initial step, we can use the output tables already created to get the taxonomic composition of our samples. First we need to gather all the output MetaPhlAn2 results per sample into a single directory and then merge them into a single table using MetaPhlAn2’s merge_metaphlan_tables.py command. After this file is created we can fix the header so that each column corresponds to a sample name without the trailing “_metaphlan_bugs_list” description. Note that MetaPhlAn2 works best for well-characterized environments, like the human gut, and has low sensitivity in other environments.
这是那篇原始的文章:Arumugam, M., Raes, J., et al. (2011) Enterotypes of the human gut microbiome, Nature,doi://10.1038/nature09944 在谷歌上一搜,作者竟然做了个分析肠型的教程在这,学习一下:http://enterotyping.embl.de/enterotypes.html 这是2018年大佬们的共识文章:这是国人翻译的这篇文章,http://blog.sciencenet.cn/blog-3334560-1096828.html 当然,如果你只需要获得自己的结果或者自己课题的结果,不需要跑代码的,有最新的网页版分型,更好用,网址也放在这,同样也是上面翻译的那篇文章里提到的网址:http://enterotypes.org/ 只需要把菌属的含量比例文件上就能很快得到结果。
This is a somewhat specialized problem that forms part of a lot of data science and clustering workflows. It starts with a relatively straightforward question: if we have a bunch of measurements for two different things, how do we come up with a single number that represents the difference between the two things?
An example will make the question clearer. Let’s load our olympic medal dataset:
import pandas as pd
pd.options.display.max_rows = 10
pd.options.display.max_columns = 6
data = pd.read_csv("https://raw.githubusercontent.com/mojones/binders/master/olympics.csv", sep="\t")
data
City
Year
Sport
…
Medal
Country
Int Olympic Committee code
0
Athens
1896
Aquatics
…
Gold
Hungary
HUN
1
Athens
1896
Aquatics
…
Silver
Austria
AUT
2
Athens
1896
Aquatics
…
Bronze
Greece
GRE
3
Athens
1896
Aquatics
…
Gold
Greece
GRE
4
Athens
1896
Aquatics
…
Silver
Greece
GRE
…
…
…
…
…
…
…
…
29211
Beijing
2008
Wrestling
…
Silver
Germany
GER
29212
Beijing
2008
Wrestling
…
Bronze
Lithuania
LTU
29213
Beijing
2008
Wrestling
…
Bronze
Armenia
ARM
29214
Beijing
2008
Wrestling
…
Gold
Cuba
CUB
29215
Beijing
2008
Wrestling
…
Silver
Russia
RUS
29216 rows × 12 columns
and measure, for each different country, the number of medals they’ve won in each different sport:
Each country has 42 columns giving the total number of medals won in each sport. Our job is to come up with a single number that summarizes how different those two lists of numbers are. Mathematicians have figured out lots of different ways of doing that, many of which are implemented in the scipy.spatial.distance module.
If we just import pdist from the module, and pass in our dataframe of two countries, we’ll get a measuremnt:
from scipy.spatial.distance import pdist
pdist(summary.loc[['Germany', 'Italy']])
array([342.3024978])
That’s the distance score using the default metric, which is called the euclidian distance. Think of it as the straight line distance between the two points in space defined by the two lists of 42 numbers.
Now, what happens if we pass in a dataframe with three countries?
But it’s not easy to figure out which belongs to which. Happily, scipy also has a helper function that will take this list of numbers and turn it back into a square matrix:
from scipy.spatial.distance import squareform
squareform(pdist(summary.loc[['Germany', 'Italy', 'France']]))
Hopefully this agrees with our intuition; the numbers on the diagonal are all zero, because each country is identical to itself, and the numbers above and below are mirror images, because the distance between Germany and France is the same as the distance between France and Germany (remember that we are talking about distance in terms of their medal totals, not geographical distance!)
Finally, to get pairwise measurements for the whole input dataframe, we just pass in the complete object and get the country names from the index:
A nice way to visualize these is with a heatmap. 137 countries is a bit too much to show on a webpage, so let’s restrict it to just the countries that have scored at least 500 medals total:
import seaborn as sns
import matplotlib.pyplot as plt
# make summary table for just top countries
top_countries = (
data
.groupby('Country')
.filter(lambda x : len(x) > 500)
.groupby(['Country', 'Sport'])
.size()
.unstack()
.fillna(0)
)
# make pairwise distance matrix
pairwise_top = pd.DataFrame(
squareform(pdist(top_countries)),
columns = top_countries.index,
index = top_countries.index
)
# plot it with seaborn
plt.figure(figsize=(10,10))
sns.heatmap(
pairwise_top,
cmap='OrRd',
linewidth=1
)
Now that we have a plot to look at, we can see a problem with the distance metric we’re using. The US has won so many more medals than other countries that it distorts the measurement. And if we think about it, what we’re really interested in is not the exact number of medals in each category, but the relative number. In other words, we want two contries to be considered similar if they both have about twice as many medals in boxing as athletics, for example, regardless of the exact numbers.
Luckily for us, there is a distance measure already implemented in scipy that has that property – it’s called cosine distance. Think of it as a measurement that only looks at the relationships between the 44 numbers for each country, not their magnitude. We can switch to cosine distance by specifying the metric keyword argument in pdist:
# make pairwise distance matrix
pairwise_top = pd.DataFrame(
squareform(pdist(top_countries, metric='cosine')),
columns = top_countries.index,
index = top_countries.index
)
# plot it with seaborn
plt.figure(figsize=(10,10))
sns.heatmap(
pairwise_top,
cmap='OrRd',
linewidth=1
)
And as you can see we spot some much more interstesting patterns. Notice, for example, that Russia and Soviet Union have a very low distance (i.e. their medal distributions are very similar).
When looking at data like this, remember that the shade of each cell is not telling us anything about how many medals a country has won – simply how different or similar each country is to each other. Compare the above heatmap with this one which displays the proportion of medals in each sport per country:
plt.figure(figsize=(10,10))
sns.heatmap(
top_countries.apply(lambda x : x / x.sum(), axis=1),
cmap='BuPu',
square=True,
cbar_kws = {'fraction' : 0.02}
)
Finally, how might we find pairs of countries that have very similar medal distributions (i.e. very low numbers in the pairwise table)? By far the easiest way is to start of by reshaping the table into long form, so that each comparison is on a separate row:
# create our pairwise distance matrix
pairwise = pd.DataFrame(
squareform(pdist(summary, metric='cosine')),
columns = summary.index,
index = summary.index
)
# move to long form
long_form = pairwise.unstack()
# rename columns and turn into a dataframe
long_form.index.rename(['Country A', 'Country B'], inplace=True)
long_form = long_form.to_frame('cosine distance').reset_index()
Now we can write our filter as normal, remembering to filter out the unintersting rows that tell us a country’s distance from itself!
This is the complete workflow used to generate a random forest model using output data from an hmmsearch of your protein coding genes against eggNOG gamma proteobacterial protein HMMs. Before running this notebook, run the parse_hmmsearch.pl script to get a tab-delimited file containing bitscores for all isolates.
“`{r, read in data}
# library(gplots)
library(caret)
library(randomForest)
set.seed(1)
# set the directory you are working from
directory <- “/home/shenzy/UNISED/CII_ECOR_update_final_164genes”
# Reading in the eggNOG model scores, checking to make sure that top eggNOG model hit for each protein in the orthogroup is the same
traindata <- read.delim(paste(directory, “/bitscores.tsv”, sep=””))
traindata <- t(traindata)
phenotype <- read.delim(paste(directory, “/phenotype.tsv”, sep=””), header=F)
phenotype[,1] <- make.names(phenotype[,1])
traindata <- cbind.data.frame(traindata, phenotype=phenotype[match(row.names(traindata), phenotype[,1]),2])
traindata[is.na(traindata)] <- 0
# traindata <- na.roughfix(traindata)
traindata <- traindata[,-nearZeroVar(traindata)]
names(traindata) <- make.names(names(traindata))
“`
The following section is an optional step for picking the best values of mtry (number of gnees sampled per node) and ntree (number of trees in your random forest) for building your model. Instead of running all of the code at once, proceed through each step or model building and examine the figures produced. These will give you an indication of the point at which the performance of your model starts to level off.
In general, greater values of ntree and mtry will give you better stability in the top genes that are identified by the model. You can alternatively skip this step and proceed immediately to the next one, where values of 10,000 trees and p/10 genes per node (where p is total number of genes in the training data) have been chosen as a good starting point.
This section allows you to build a model through iterative feature selection, using parameters that we feel are sensible. You can substitute in your own parameters chosen from the process above if you prefer.
The following section will show you how the performance of your model has improved as you iterated through cycles of feature selection. It will give you an idea of whether you have performed enough cycles, or whether you need to carry on.
You can look at both the out-of-bag votes, which are just votes cast by trees on data they were not trained on. This gives you a good idea of how the model would score strains that are distantly related to your training data. The second plot shows you votes cast by all of the trees on all of the serovars, and gives you a better idea of how your model would scores similar strains.
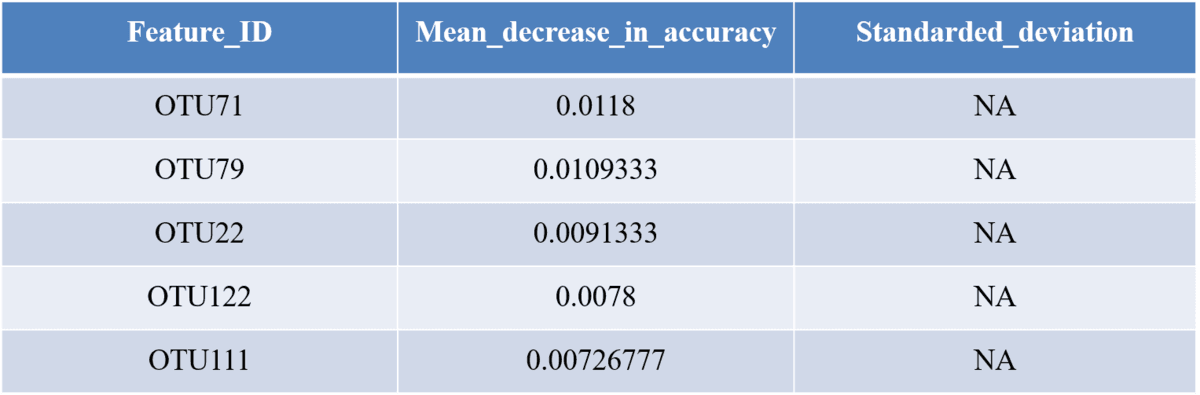
This section shows you how frequently particular genes are identified as top predictors across different iterations of model building, and how much importance they are assigned. Because model building involved a lot of stochasticity, you may find that your top predictors are quite subject to change across iterations.
“`{r, assessing stability of predictors}
set.seed(1)
usefulgenes <- data.frame()
topgenes <- data.frame()
for(i in 1:10) {
model <- randomForest(phenotype ~ ., data=traindata, ntree=10000, mtry=param/10, na.action=na.roughfix)
usefulgenes <- rbind(usefulgenes, cbind(model=i, gene=names(model$importance[model$importance>0,]), model$importance[model$importance>0]))
topgenes <- rbind(topgenes, cbind(model=i, gene=names(model$importance[order(model$importance, decreasing=T),][1:20]), model$importance[order(model$importance, decreasing=T),][1:20]))
}
png(paste(directory, “/gene_usefulnessnew.png”, sep=””))
hist(table(usefulgenes$gene), col=”grey”, main=””, xlab=”Number of times each gene was useful (VI >) in a model”)
dev.off()
sum(table(usefulgenes$gene)==10)
sum(table(usefulgenes$gene)<10)
topgenes$V3 <- as.numeric(as.character(topgenes$V3))
ggplot(topgenes, aes(x=model, y=gene, fill=as.numeric(V3))) + geom_tile() + ggtitle(“Imporance values for top genes across model iterations”) + scale_fill_continuous(“Importance”)
table(topgenes$gene)
png(paste(directory, “/topgenesnew.png”, sep=””))
hist(table(topgenes$gene), col=”grey”, main=””, xlab=”Number of times each gene appeared in the\ntop 20 predictors”)
dev.off()
save(usefulgenes, topgenes, file=paste(directory, “/allgenemodels.Rdata”, sep=””))
“`
This section allows you to look at the COG categories of your top predictor genes, compared to the COG categories in your original training set, to see if any are over-represented.
“`{r, COG analysis}
annotations <- read.delim(paste(directory, “/gproNOG.annotations.tsv”, sep=””), header=F) # matcing annotation file for your chosen model set can be downloaded from http://eggnogdb.embl.de/#/app/downloads
nogs <- read.delim(paste(directory, “/models_used_20ref.tsv”, sep=””), header=F)
nogs[,2] <- sub(“.*NOG\\.”, “”, nogs[,2])
nogs[,2] <- sub(“.meta_raw”, “”, nogs[,2])
info <- annotations[match(nogs[,2], annotations[,2]),]
info$V5 <- as.character(info$V5)
# proportion of each COG catergory that comes up in the top indicators
background <- nogs[match(colnames(traindata), make.names(nogs[,1])),2]
background_COGs <- annotations[match(background, annotations[,2]),5]
library(plyr)
bg_COGs2 <- aaply(as.character(background_COGs), 1, function(i){
if(!is.na(i)) {
if(nchar(i)>1) {
char <- substr(i,1,1)
for(n in 2:nchar(i)) {
char <- paste(char,substr(i,n,n),sep=”.”)
}
return(char)
} else {
return(i)
}
} else{
return(NA)
}
})
cmodel6$predicted
names(cmodel6$importance[order(cmodel6$importance[,1], decreasing=T),])[1:10]
# compare votes and phenotypes for the control and real datasets
cbind(cmodel6$votes, control$phenotype, model6$votes, train6$phenotype)
“`
This approach repeats the full model-building process 5 times to see how similar the top predictor genes are.
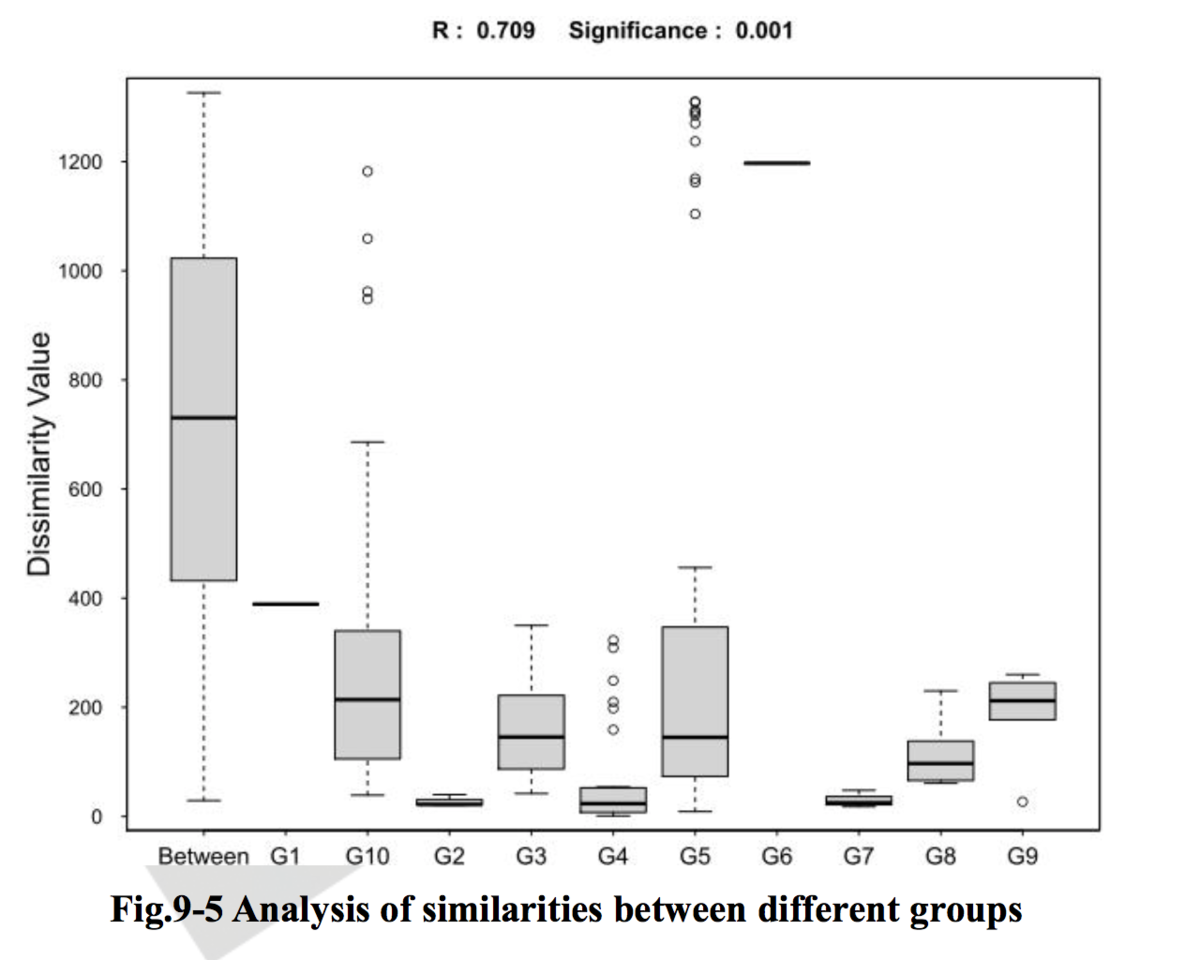
注:理论上,R值范围为-1到+1,实际中R值一般从0到1,R值接近1表示组间差异越大于组内差异,R值接近0则表示组间和组内没有明显差异。P值则反映了分析结果的统计学显著性,P值越小,表明各样本分组之间的差异显著性越高,P< 0.05表示统计具有显著性;Number of permutation表示置换次数。
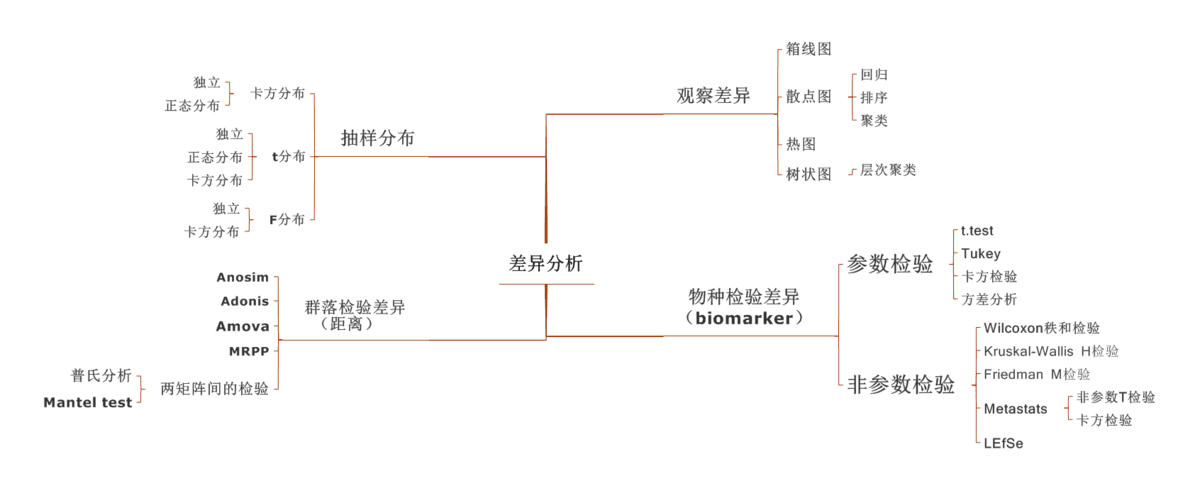
相似性分析(ANOSIM)是一种非参数检验,用来检验组间(两组或多组)的差异是否显著大于组内差异,从而判断分组是否有意义。首先利用 Bray-Curtis 算法计算两两样品间的距离,然后将所有距离从小到大进行排序, 按以下公式计算 R 值,之后将样品进行置换,重新计算 R值,R大于 R 的概率即为 P 值。
Sylvain I A, Adams R I, Taylor J W. A different suite: The assemblage of distinct fungal communities in water-damaged units of a poorly-maintained public housing building[J]. PloS one, 2019, 14(3): e0213355.
其中,
r0= mean rank of between group dissimilarities 即组间差异性秩的平均值
rw= mean rank of within group dissimilarities 即组内差异性秩的平均值
n=the number of samples 即样本总数量
➜ docker-compose up
Starting test_caddy_1 ... done
Attaching to test_caddy_1
caddy_1 | Activating privacy features...
caddy_1 |
caddy_1 | Your sites will be served over HTTPS automatically using Let's Encrypt.
caddy_1 | By continuing, you agree to the Let's Encrypt Subscriber Agreement at:
caddy_1 | https://letsencrypt.org/documents/LE-SA-v1.2-November-15-2017.pdf
caddy_1 | Please enter your email address to signify agreement and to be notified
caddy_1 |incase of issues. You can leave it blank, but we don't recommend it.
caddy_1 | Email address: 2020/01/10 13:54:58 [INFO] acme: Registering account for
caddy_1 | 2020/01/10 13:54:58 [INFO][xx.xx.xx] acme: Obtaining bundled SAN certificate
caddy_1 | 2020/01/10 13:54:58 [INFO][xx.xx.xx] AuthURL: https://acme-v02.api.letsencrypt.org/acme/authz-v3/xxxxxx
caddy_1 | 2020/01/10 13:54:58 [INFO][xx.xx.xx] acme: Trying to solve HTTP-01
caddy_1 | 2020/01/10 13:54:58 [INFO][xx.xx.xx] Served key authentication
caddy_1 | 2020/01/10 13:54:58 [INFO][xx.xx.xx] Served key authentication
caddy_1 | 2020/01/10 13:55:03 [INFO][xx.xx.xx] The server validated our request
caddy_1 | 2020/01/10 13:55:03 [INFO][xx.xx.xx] acme: Validations succeeded; requesting certificates
caddy_1 | 2020/01/10 13:55:04 [INFO][xx.xx.xx] Server responded with a certificate.
caddy_1 | 2020/01/10 13:55:04 [INFO][xx.xx.xx] Certificate written to disk: /root/.caddy/acme/acme-v02.api.letsencrypt.org/sites/xx.xx.xx/xx.xx.xx.crt
caddy_1 | done.
Starting test_trojan_1 ... done
Attaching to test_caddy_1, test_trojan_1
trojan_1 | ssl certs is empty - checking...
trojan_1 | ssl certs is empty - checking...
trojan_1 | Welcome to trojan 1.14.0
trojan_1 |[2020-01-10 15:40:58][WARN] trojan service(server) started at 0.0.0.0:443


































Recent Comments