[Admin.DESKTOP-7JT504C] ➤ rsync -P –rsh=ssh /drives/d/Kraken_12.tar.gz cityu_jhli_1@172.16.22.11:/BIGDATA1/cityu_jhli_1/mhyleung/database/findfungi Warning: Permanently added ‘172.16.22.11’ (RSA) to the list of known hosts.
if (!require(“gplots”)) {
install.packages(“gplots”, dependencies = TRUE)
library(gplots)
}
if (!require(“RColorBrewer”)) {
install.packages(“RColorBrewer”, dependencies = TRUE)
library(RColorBrewer)
}
#########################################################
### B) Reading in data and transform it into matrix format
#########################################################
setwd(“/home/zyshen/Downloads/酵母代谢”)
data <- read.csv(“W29-knockout-0.1-SD-10X-internal.val.filter.csv”, comment.char=”#”, header=T)
rnames <- data[,1] # assign labels in column 1 to “rnames”
mat_data <- data.matrix(data[,2:ncol(data)]) # transform column 2-5 into a matrix
rownames(mat_data) <- rnames # assign row names
my_palette <- colorRampPalette(c(“red”, “yellow”, “green”))(n = 299)
# (optional) defines the color breaks manually for a “skewed” color transition
col_breaks = c(seq(-50,0,length=100), # for red
seq(0.1,20,length=100), # for yellow
seq(21,100,length=100)) # for green
heatmap.2(mat_data,
cellnote = mat_data, # same data set for cell labels
main = “Correlation”, # heat map title
notecol=”black”, # change font color of cell labels to black
density.info=”none”, # turns off density plot inside color legend
trace=”none”, # turns off trace lines inside the heat map
margins =c(8,20), # widens margins around plot
col=my_palette, # use on color palette defined earlier
breaks=col_breaks, # enable color transition at specified limits
dendrogram=”row”, # only draw a row dendrogram
Colv=”NA”)
The invsimpson calculator is the inverse of the classical Simpson diversity estimator. This parameter is preferred to other measures of alpha-diversity because it is an indication of the richness in a community with uniform evenness that would have the same level of diversity.
https://www.mothur.org/wiki/Invsimpson
Biological diversity - the great variety of life !
ClinVAP: A reporting strategy from variants to therapeutic options
Abstract
Motivation
Next-generation sequencing (NGS) has become routine in oncology and opens up new avenues of therapies, particularly in personalized oncology setting. An increasing number of cases also implies a need for a more robust, automated, and reproducible processing of long lists of variants for cancer diagnosis and therapy. While solutions for the large-scale analysis of somatic variants have been implemented, existing solutions often have issues with reproducibility, scalability, and interoperability.
Results
ClinVAP is an automated pipeline which annotates, filters, and prioritizes somatic single nucleotide variants (SNVs) provided in variant call format. It augments the variant information with documented or predicted clinical effect. These annotated variants are prioritized based on driver gene status and druggability. ClinVAP is available as a fully containerized, self-contained pipeline maximizing reproducibility and scalability allowing the analysis of larger scale data. The resulting JSON-based report is suited for automated downstream processing, but ClinVAP can also automatically render the information into a user-defined template to yield a human-readable report.
Do groups differ in composition?
Does community structure vary among regions or over time?
Do environmental variables explain community patterns?
Which species are responsible for differences among groups?
Bird abundances from 32 different plots (rows), 12 of which have 1 tree species (DIVERSITY = M) and 20 with 4 tree species (DIVERSITY = P).
Tree composition: there are a total of 6 possible tree species (treecomp), each signified with a letter A to F. Bird abundances are totalled according to their feeding guild (columns).
setwd("/Users/colleennell/Dropbox/Projects/Mexico/R")#change to data folderbirds<-read.csv('bird_by_fg.csv')head(birds)
## DIVERSITY PLOT CA FR GR HE IN NE OM
## 1 M 3 0 0 0 0 2 0 0
## 2 M 9 0 0 2 0 6 0 4
## 3 M 12 0 0 0 0 2 0 2
## 4 M 17 0 0 0 0 7 0 4
## 5 M 20 0 0 0 0 1 0 4
## 6 M 21 0 0 3 0 14 0 7
trees<-read.csv('tree_comp.csv')head(trees)
## PLOT comp A B C D E F row col
## 1 3 D 0 0 0 1 0 0 3 1
## 2 9 A 1 0 0 0 0 0 2 2
## 3 12 E 0 0 0 0 1 0 5 2
## 4 17 F 0 0 0 0 0 1 3 3
## 5 20 A 1 0 0 0 0 0 6 3
## 6 21 B 0 1 0 0 0 0 7 3
Questions: Is C. pentandara (B) associated with variation in bird species composition? Does feeding guild composition differ between monoculture and polyculture plots?
MANOVA (Multivariate analysis of variance)
Parametric test for differences between independent groups for multiple continuous dependent variables. Like ANOVA for many response variables. Requires variables to be fewer than number of smaples.
Is C. pentandara (B) associated with variation in bird species composition? Or D & F (both Fabaceae)?
bird.matrix<-as.matrix(birds[,3:9])##response variables in a sample x species matrixtrees$B<-as.factor(trees$B)bird.manova<-manova(bird.matrix~as.factor(B), data=trees)##manova testsummary(bird.manova)
Problem: Most ecological data is overdispersed, has many 0’s or rare species, unequal sample sizes. Solution: Dissimilarity coefficients, permutation tests
PERMANOVA: Permutational multivariate analysis of variance
Non-paramentric, based on dissimilarities. Allows for partitioning of variability, similar to ANOVA, allowing for complex design (multiple factors, nested design, interactions, covariates). Uses permutation to compute F-statistic (pseudo-F). Interactive app demonstrating permutation tests
Based on Legendre & Anderson (1999, Ecological Monographs) and Anderson (2001, Austral Ecology).
Null hypothesis: Groups do not differ in spread or positioni n multivaraite space.
1. Transform or standardize data
Use square root or proportions to minimize influence of most abundant groups.
##
## Permutation test for homogeneity of multivariate dispersions
## Permutation: free
## Number of permutations: 999
##
## Response: Distances
## Df Sum Sq Mean Sq F N.Perm Pr(>F)
## Groups 1 0.00369 0.0036924 0.2231 999 0.638
## Residuals 30 0.49659 0.0165530
Non-metric multi-dimensional scaling. Unconstrained ordination. See (https://jonlefcheck.net/2012/10/24/nmds-tutorial-in-r/).
The goal of NMDS is to represent the original position of communities in multidimensional space as accurately as possible using a reduced number of dimensions that can be easily visualized. NMDS uses rank orders to preserve distances among objects thus can accomodate a variety of data types.
Configure samples in 2-dimensional space:
birdMDS<-metaMDS(bird.mat, distance="bray", k=2, trymax=35, autotransform=TRUE)##k is the number of dimensionsbirdMDS##metaMDS takes eaither a distance matrix or your community matrix (then requires method for 'distance=')stressplot(birdMDS)
Stress: similarity of observed distance to ordination distance. < 0.15 to indidates acceptable fit.
install.packages('ggplot2')##plotting packagelibrary(ggplot2)##pull points from MDSNMDS1<-birdMDS$points[,1]##also found using scores(birdMDS)NMDS2<-birdMDS$points[,2]bird.plot<-cbind(birds, NMDS1, NMDS2, trees)#plot ordinationp<-ggplot(bird.plot, aes(NMDS1, NMDS2, color=DIVERSITY))+geom_point(position=position_jitter(.1), shape=3)+##separates overlapping pointsstat_ellipse(type='t',size=1)+##draws 95% confidence interval ellipsestheme_minimal()p
ordisurf(birdMDS, bird.mat[,'IN'], bubble=TRUE)##bubble size reflects abundance of insectivores
##
## Family: gaussian
## Link function: identity
##
## Formula:
## y ~ s(x1, x2, k = 10, bs = "tp", fx = FALSE)
##
## Estimated degrees of freedom:
## 5.6 total = 6.6
##
## REML score: 35.72947
Resources: GUSTA ME – Provides several ‘wizards’ in choosing the correct statistical test, walkthrough examples of multivariate analyses, and guide to the major types of analyses.
#!/usr/bin/env python
import os,re,sys,string,commands,getopt,subprocess,glob,csv
import prettytable as pt
from os import path
#SL335752_kneaddata_paired_1_kneaddata_paired_1.fastq.gz_rep.mpa.txt_bracken.txt
#SL311013_1_kneaddata_paired_1_kneaddata_paired_1.fastq.gz_rep_mpa.txt_bracken.txt
def main():
dic = {}
unique_speciesname = []
speciesname = []
samplenames = []
for d in os.listdir(‘.’):
print(d)
a = []
a = re.split(“_kneaddata_paired_1_”,d)
if len(a) >=2:
samplename = a[0]
dic[a[0]] = {}
#os.rename(d, newname)
fh = open(d, ‘r’)
fhlines = fh.readlines()
fh.close()
for line in fhlines:
line = line.strip()
if re.search(“name”, line):
continue
else:
b = []
b = re.split(“\t”,line)
speciesname.append(b[0])
length = len(b)
dic[a[0]][b[0]] = b[length-1]
unique = [unique_speciesname.append(x) for x in speciesname if x not in unique_speciesname]
for sample_name in dic.keys():
samplenames.append(sample_name)
for name in unique_speciesname:
#print name
if dic[sample_name].has_key(name):
print_line = name + “\t” + dic[sample_name][name]
else:
dic[sample_name][name] = “0”
print_line = name + “\t” + dic[sample_name][name]
#print dict_data_mine
try:
with open(csv_file, ‘w’) as csvfile:
writer = csv.DictWriter(csvfile, fieldnames=csv_columns)
writer.writeheader()
for data in dict_data_mine:
writer.writerow(data)
except IOError:
print(“I/O error”)
if __name__== ‘__main__':
main()
#!/usr/bin/env python
import os,re,sys,string,commands,getopt,subprocess,glob
from os import path
#/disk/rdisk08/mhyleung/loreal_shotgun2/NovaSeq_new/decontam_output_genome/paired_completed/szy_test
#LOR294C_S130_R1_001_kneaddata_paired_1_decontaminated.fastq.paired.fq.gz
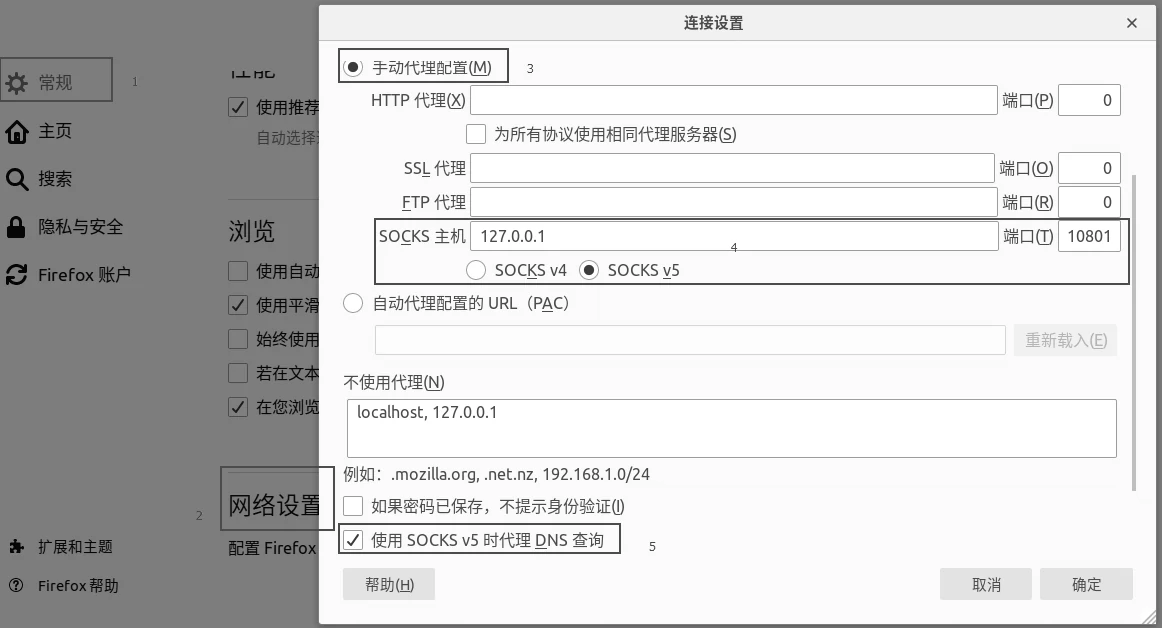
root@vm:~# service v2ray stop
root@vm:~# service v2ray start
root@vm:~# service v2ray status
● v2ray.service - V2Ray Service
Loaded: loaded (/etc/systemd/system/v2ray.service; enabled; vendor preset: en
Active: active (running) since Sat 2019-05-18 08:58:43 CST; 5s ago
Main PID: 8025 (v2ray)
Tasks: 7 (limit: 2311)
CGroup: /system.slice/v2ray.service
└─8025 /usr/bin/v2ray/v2ray -config /etc/v2ray/config.json
5月 18 08:58:43 vm systemd[1]: Started V2Ray Service.
5月 18 08:58:43 vm v2ray[8025]: V2Ray 4.18.0 (Po) 20190228
5月 18 08:58:43 vm v2ray[8025]: A unified platform for anti-censorship.
5月 18 08:58:44 vm v2ray[8025]: 2019/05/18 08:58:44 [Warning] v2ray.com/core: V2





Recent Comments